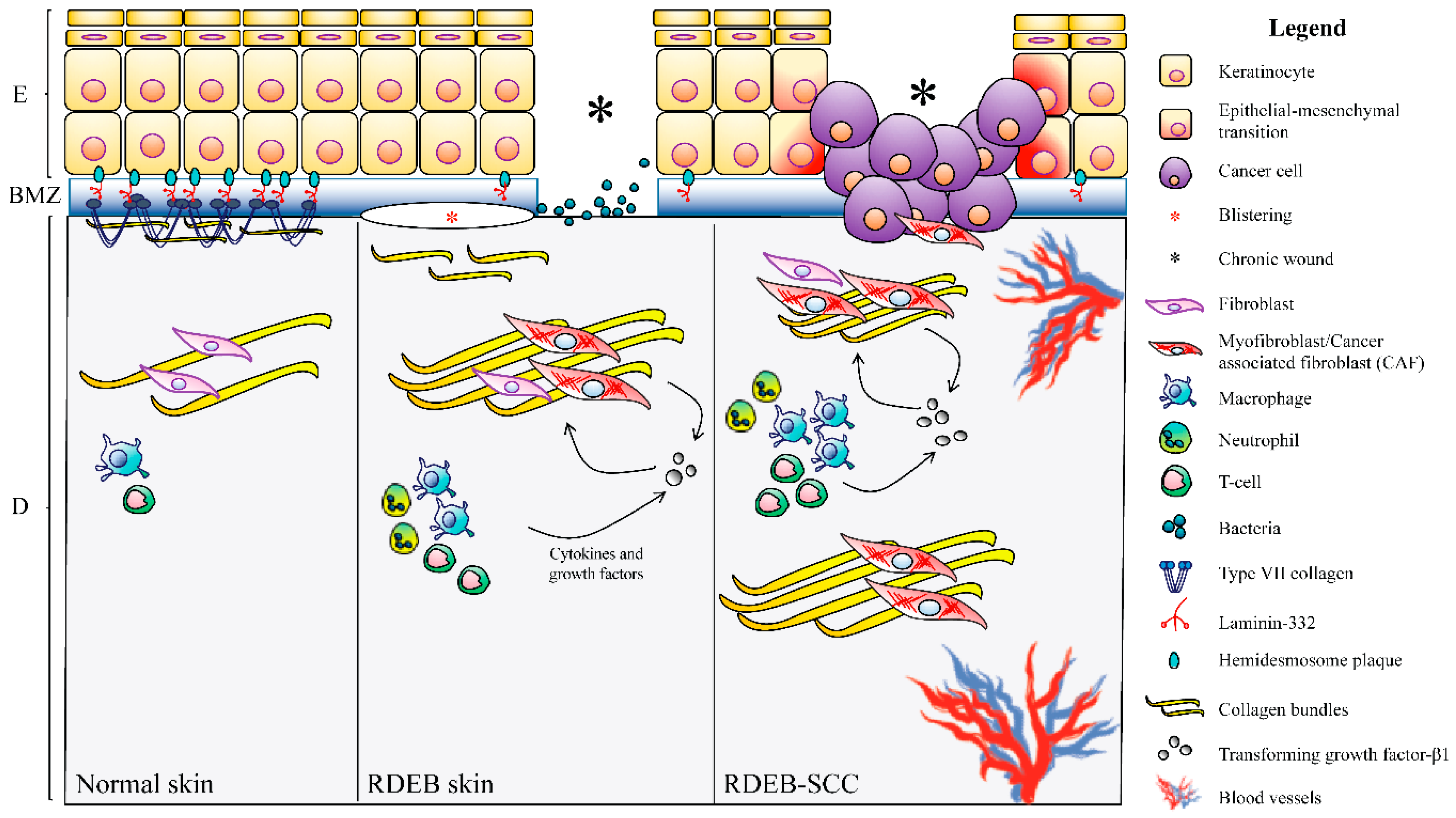
Pathomechanisms of Altered Wound Healing in Recessive Dystrophic
IJMS Free FullText Epidermolysis BullosaAssociated Squamous Cell
In October 2003 the German network of excellence "Epidermolysis bullosa: molecular pathomechanisms and novel therapeutic approaches" initiated its activities. The network partners are physicians and scientists working on epidermolysis bullosa (EB), basement membranes and structural proteins. The cli.
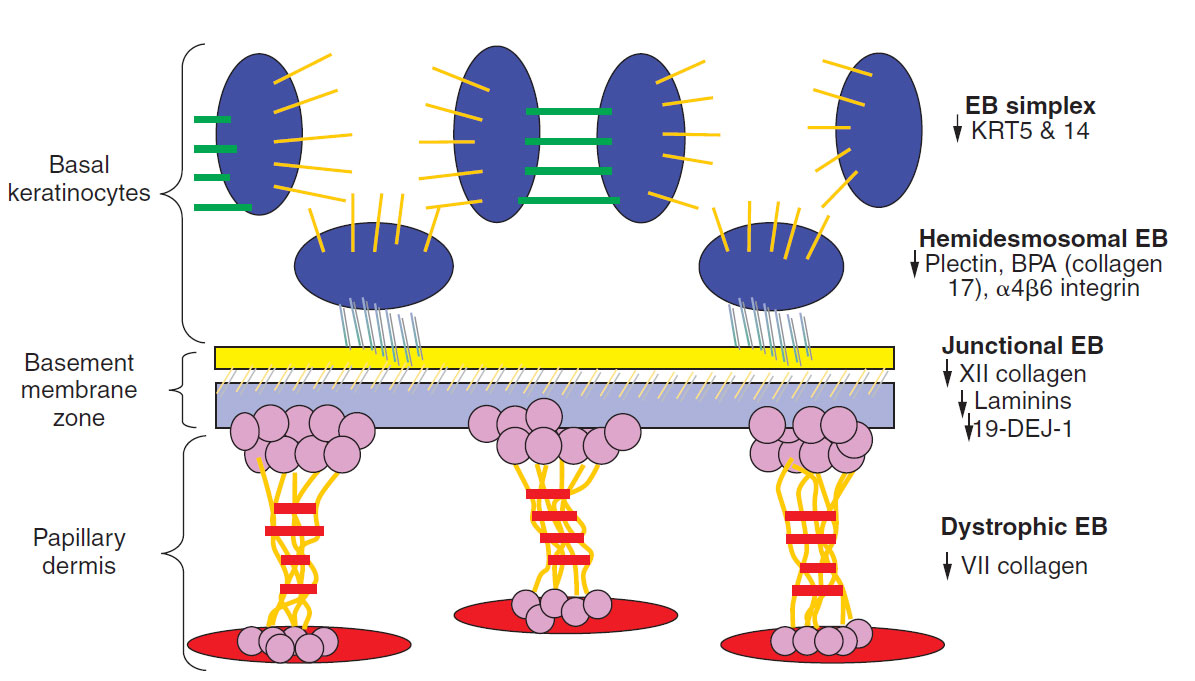
What structural components of the epidermis are involved in blistering
Hereditary epidermolysis bullosa Summary The term epidermolysis bullosa (EB) includes a group of rare genodermatoses cha-racterized by mutational impairment of the structural and functional integrity of in-traepidermal adhesion and dermoepidermal anchorage. Clinically, these disorders

Pathomechanisms of Altered Wound Healing in Recessive Dystrophic
New insights into molecular genetics and pathomechanisms in epidermolysis bullosa (EB), methodological and technological advances in molecular biology as well as designated funding initiatives and facilitated approval procedures for orphan drugs have boosted translational research perspectives for this devastating disease. This is echoed by the increasing number of clinical trials assessing.

Classification and molecular characteristics of epidermolysis bullosa
Inherited epidermolysis bullosa (EB) is a group of genetic diseases associated with skin fragility, which leads to the formation of blisters, erosions, and scars on the skin and mucous membranes in response to minimal mechanical trauma. 1 EB is clinically and genetically very heterogeneous, comprising phenotypes with contrasting levels of.

Clinical practice guidelines for laboratory diagnosis of epidermolysis
Abstract. Epidermolysis bullosa (EB), a phenotypically and genetically heterogeneous disorder, has been linked to mutations in the genes encoding structural proteins that reinforce skin integrity via dermal-epidermal adhesion. Breakdowns in these adhesion mechanisms result in four different subtypes of EB classified on the basis of the level of.

Epidermolysis bullosa
The network partners are physicians and scientists working on epidermolysis bullosa (EB), basement membranes and structural proteins. The clinical partners and associated specialists improve interdisciplinary management of patients with EB and coordinate diagnostic and therapeutic procedures.
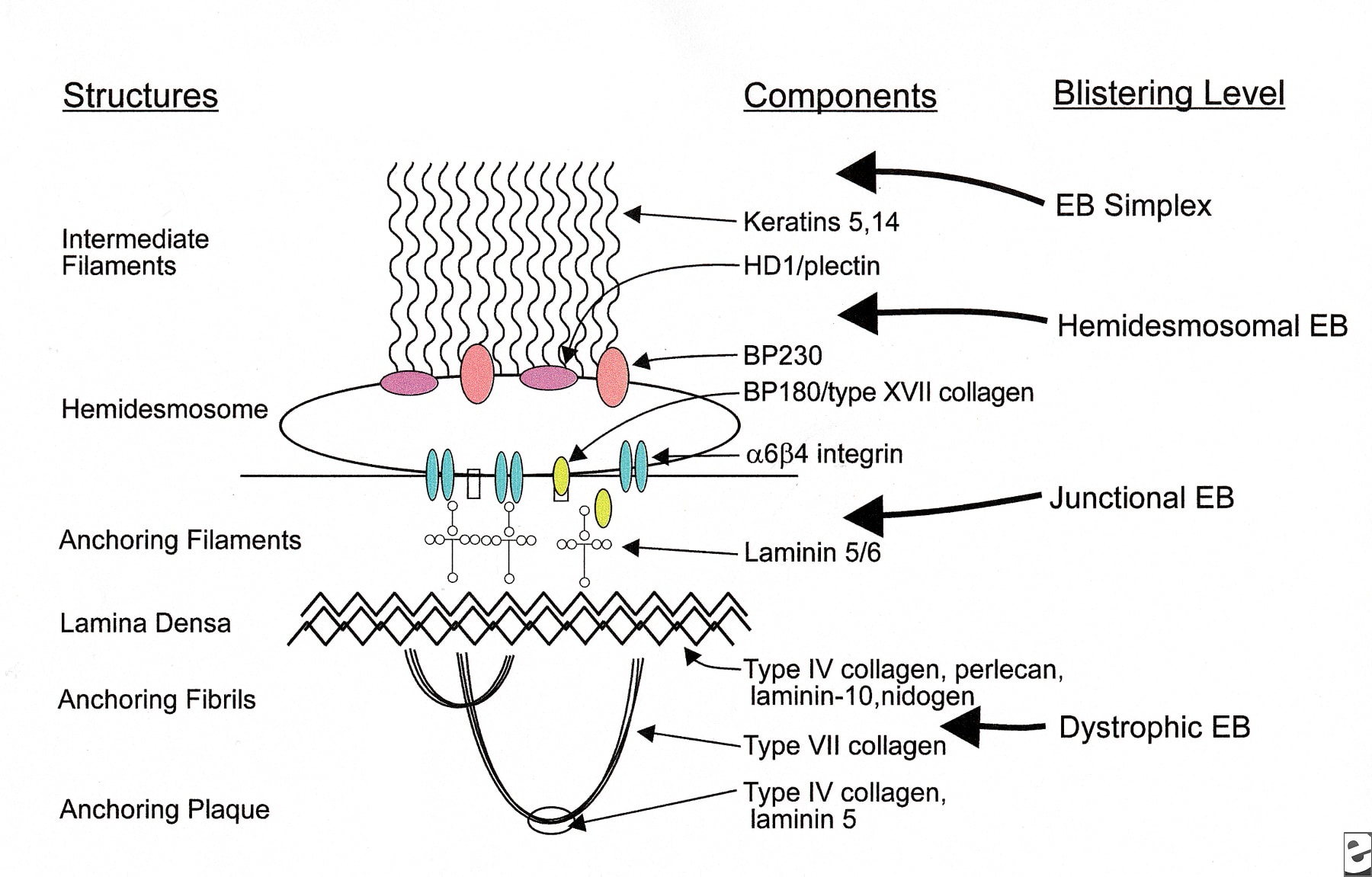
Pathology Outlines Epidermolysis bullosa
This website requires cookies, and the limited processing of your personal data in order to function. By using the site you are agreeing to this as outlined in our privacy notice and cookie policy.privacy notice and cookie policy.
.png/800px-Epidermolysis_bullosa_(2).png)
Epidermolysis Bullosa Physiopedia
New insights into molecular genetics and pathomechanisms in epidermolysis bullosa (EB), methodological and technological advances in molecular biology as well as designated funding initiatives and facilitated approval procedures for orphan drugs have boosted translational research perspectives for this devastating disease. This is echoed by the.

Diagnostics Free FullText Epidermolysis Bullosa—A Different
structural and functional integrity of the epidermis and dermo-epidermal junction underlie the four main types of epidermolysis bullosa (EB), featuring skin blistering within the epidermis (EB simplex), the lamina lucida (junctional EB), the upper dermis (DEB) or at mixed levels (Kindler EB). Letters in boxes next to the protein names indicate
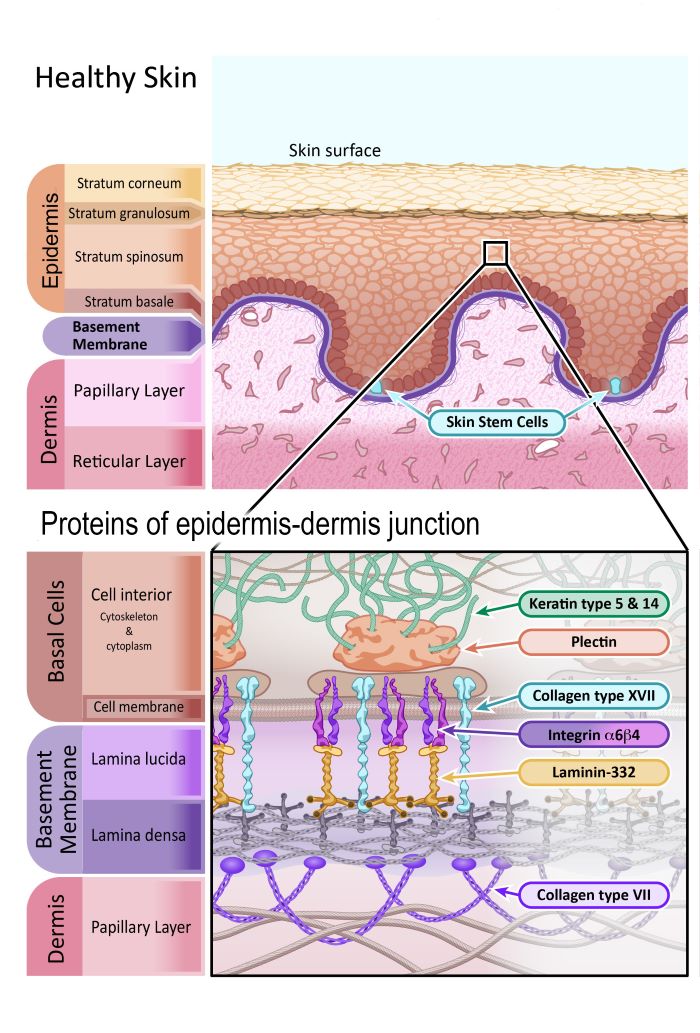
Epidermolysis Bullosa How could gene and cell therapy help? EuroGCT
Epidermolysis bullosa (EB) comprises a group of rare skin fragility disorders characterised by mutations in basement membrane zone (BMZ) structural proteins in the skin [].To date, 16 different genes have been implicated across the four classical EB types, EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler EB [].These types and their underlying subtypes are classified based.

Classification and molecular characteristics of epidermolysis bullosa
Abstract. Epidermolysis bullosa (EB) is the umbrella term for a group of rare inherited skin fragility disorders caused by mutations in at least 20 different genes. There is no cure for any of the subtypes of EB resulting from different mutations, and current therapy only focuses on the management of wounds and pain.

Figure 2 from Ultrastructure and molecular pathogenesis of
Epidermolysis bullosa (EB) is an inherited, heterogeneous group of rare genetic dermatoses characterized by mucocutaneous fragility and blister formation, inducible by often minimal trauma. A.

Toward treatment and cure of epidermolysis bullosa PNAS
Introduction. Epidermolysis bullosa (EB) is a genetically heterogenous group of rare disorders that causes mechanical fragility and blistering of the skin and in some cases the epithelial lining of other organs, in response to minimal or no apparent trauma. 1 EB affects approximately 1 in 17,000 live births, with 500,000 cases estimated worldwide. 2 EB can be classified into 4 main types based.
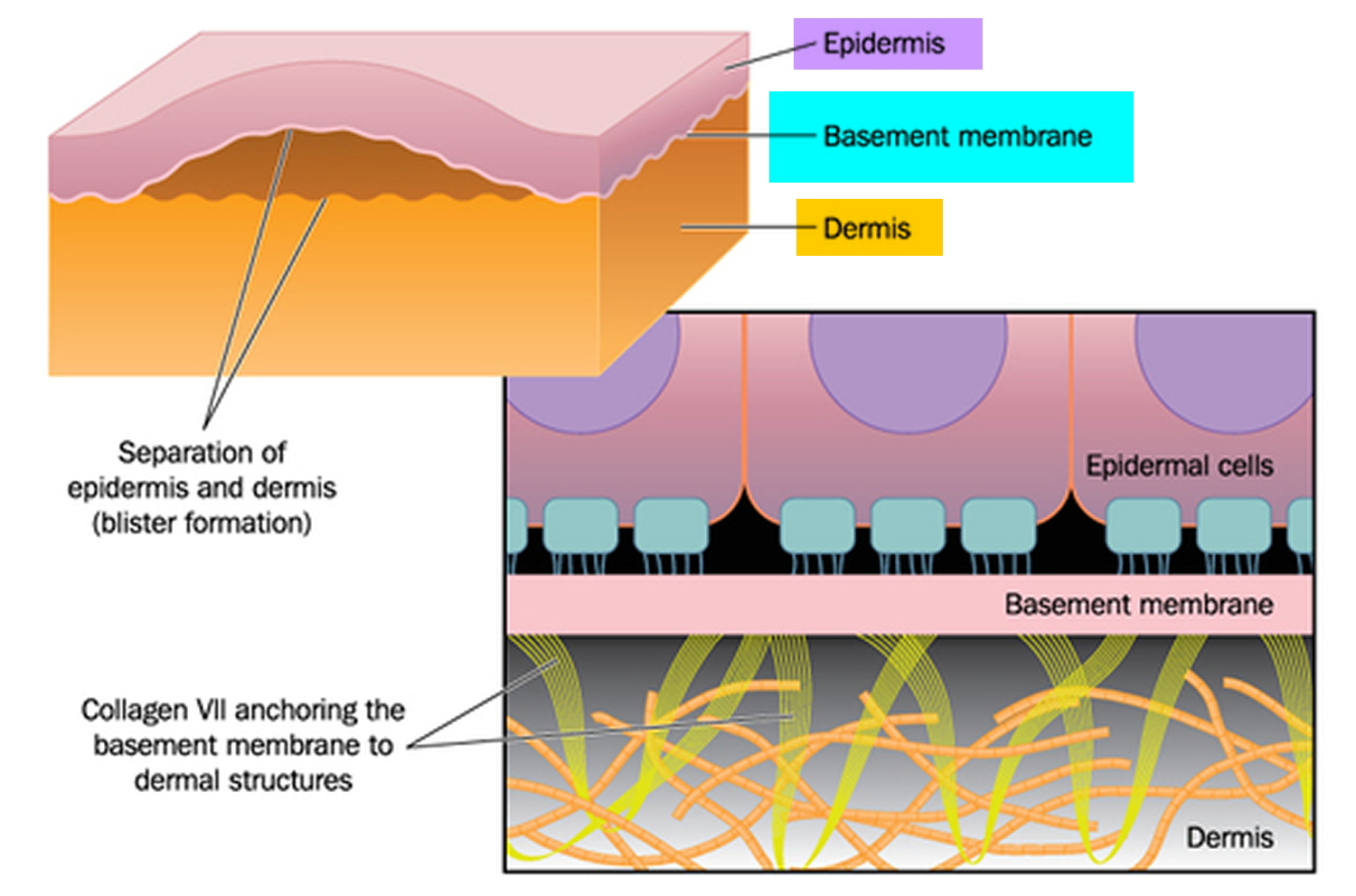
Epidermolysis Bullosa Causes, Symptoms, Types, Prognosis & Treatment
Epidermolysis bullosa (EB), a phenotypically and genetically heterogeneous disorder, has been linked to mutations in the genes encoding structural proteins that reinforce skin integrity via dermal-epidermal adhesion. Breakdowns in these adhesion mechanisms result in four different subtypes of EB classified on the basis of the level of tissue separation within the cutaneous basement membrane.
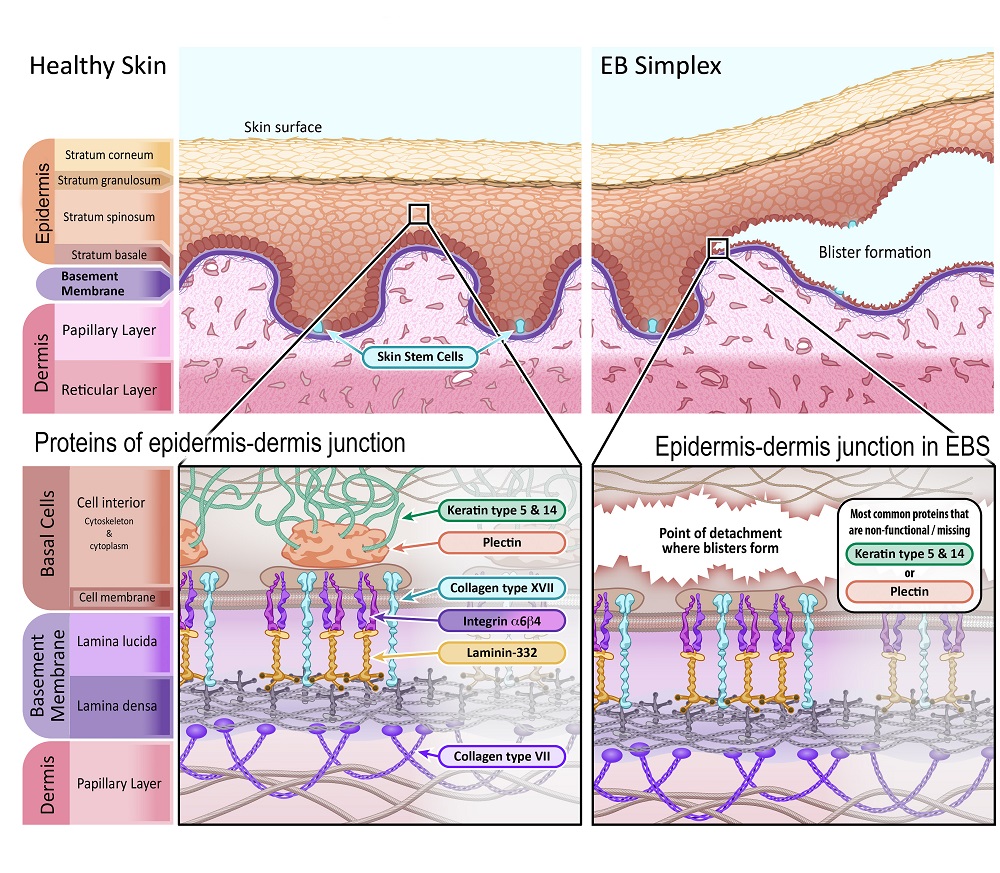
Epidermolysis Bullosa How could gene and cell therapy help? EuroGCT
DOI: 10.1016/j.matbio.2022.04.007 Corpus ID: 248489948; Pathomechanisms of epidermolysis bullosa: Beyond structural proteins. @article{Harvey2022PathomechanismsOE, title={Pathomechanisms of epidermolysis bullosa: Beyond structural proteins.}, author={Nailah Harvey and Leila Youssefian and Amir Hossein Saeidian and Hassan Vahidnezhad and Jouni Uitto}, journal={Matrix biology : journal of the.
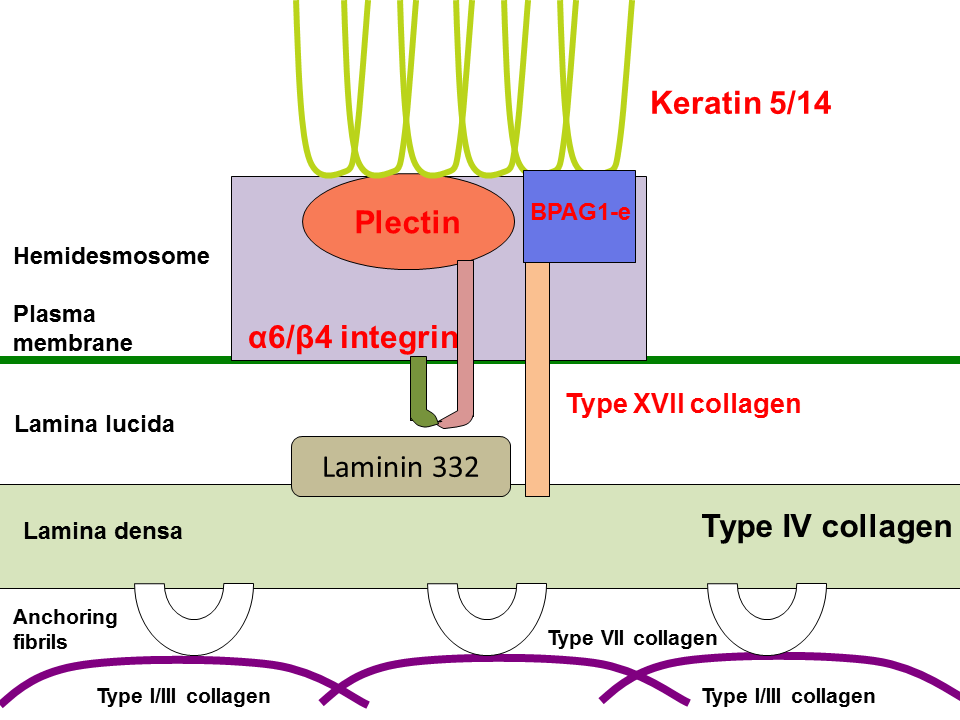
Epidermolysis Bullosa Simplex IntechOpen
Epidermolysis bullosa (EB) defines a prototypic group of rare, inherited dermatoses, characteristically featuring skin fragility secondary to structural defects in the dermo-epidermal junction. This skin fragility creates an impaired tolerance to mechanical stress. Trivial mechanical trauma and shear stress can provoke skin blistering, erosions, and ulceration.[1] This places patients with.
